
AMPK: una vista estructural a través de ChimeraX

RESUMEN
Considerando la diversidad de cada proteína dependiendo la combinación de aminoácidos en cada secuencia polipeptídica y sus campos de fuerza que formen, tendrán una estructura única, con un programa extensible para la visualización interactiva y el análisis de estructuras moleculares se van analizar la estructura de la enzima AMPK. Se eligió una proteína de interés usando bases de datos como PBD. A través de determinar distancias, campos de fuerza, aminoácidos se identifican las estructuras supersecundarias Unidades beta-alfa-beta; meandro Beta como parte de las cadenas de la AMPK.
Palabras clave: Aminoácido, estructura 3d proteína, ChimeraX, AMPK, enlaces no covalentes
AMPK: a structural view through ChimeraX
ABSTRACT
Considering the diversity of each protein depending on the combination of amino acids in each polypeptide sequence and their force fields they form, they will have a unique structure, with an extensible program for interactive visualization and analysis of molecular structures. A protein of interest was chosen from databases such as PBD. Through determining distances, force fields, amino acids are identified supersecondary structures beta-alpha-beta units; Beta meander as part of the AMPK chains.
Keywords: Amino acid, 3d protein structure, ChimeraX, AMPK, protein
INTRODUCCIÓN
Las proteínas constituyen la mayor parte de la masa seca de una célula y tienen muchas funciones, un ejemplo de ello: sirven como
moléculas de adhesión celular que unen unas células con otras y con la matriz extracelular, como hormonas que transmiten señales desde un grupo de células a otro, como canales iónicos a través de
las membranas, así como enzimas (Lieberman & Peet, 2018). Otras proteínas especializadas actúan como anticuerpos, toxinas, fibras elásticas, cuerdas o fuentes de luminiscencia (Alberts, y otros, 2015).
Las proteínas son un grupo diverso de macromoléculas, esta diversidad está directamente relacionada con la diversidad de combinación de cada monómero de los 20 aminoácidos que existen
en la biología (Mckee & Mckee, 2014); así como su estructura tridimensional. Esto depende de los tres tipos de enlaces no covalentes que participan en el plegamiento de las proteínas (Alberts, y otros, 2015).

Ilustración 1 Estructura de un aminoácido. Un aminoácido está formado por: (H2N) un extremo amino; © un carbono alfa; (CooH) extremo carboxilo (cuando se quita el Hidrógeno, queda en su forma iónica); (H) un hidrógeno y ® su cadena lateral le da identidad.
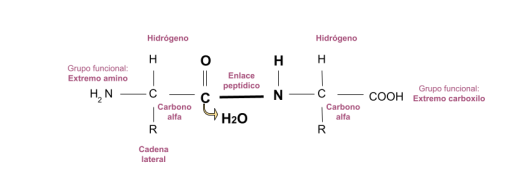
Ilustración 2 Unión de dos aminoácidos. Los aminoácidos se pueden unir entre ellos, se unen de un extremo amino con un extremo carboxilo. Se pueden pegar más aminoácidos y la orientación se mantiene (puede ser de 100, 200 hasta proteínas de 1000 aminoácidos), todas van a tener un extremo amino y un extremo carboxilo.
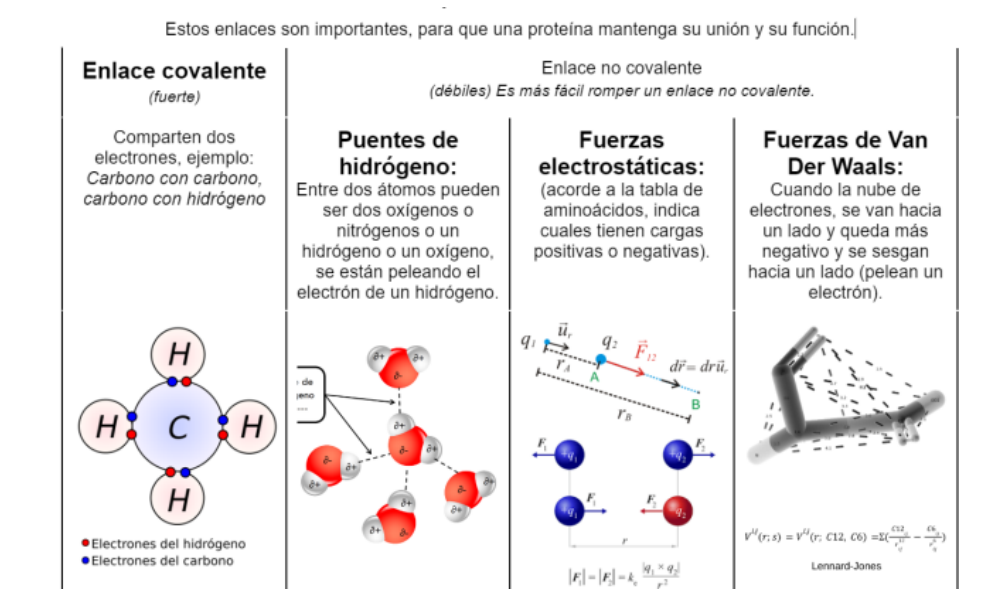
Ilustración 3 Tipos de enlace que tienen los
aminoácidos. Enlace covalente: electrones son atraídos simultáneamente por los dos núcleos atómicos. Un enlace covalente se forma cuando la diferencia entre las electronegatividades de dos
átomos es demasiado pequeña para que se produzca una
transferencia de electrones para formar iones. Enlaces no covalentes: Estos tres tipos de enlaces no covalentes participan en el plegamiento de las proteínas. Cada uno de estos enlaces por sí solos son débiles, pero muchos de ellos a la vez dan lugar a disposiciones unidas fuertemente. Como resultado de todas estas interacciones, la mayoría de proteínas tienen una estructura tridimensional particular determinada por el orden de aminoácidos de su cadena.
Los científicos distinguen cuatro niveles de organización en la estructura de una proteína, para este artículo nos centraremos en la primera y segunda estructura. En la estructura primaria cada polipéptido tiene una secuencia de aminoácidos específica; por otra parte la estructura secundaria consta de tramos de cadena polipeptídica que forman hélices α y láminas ẞ, estas dos últimas
estructuras están estabilizadas por enlaces por puente de hidrógeno entre los grupos carbonilo y grupo amino del esqueleto polipeptídico. Es importante mencionar que los enlaces peptídicos son rígidos, por lo que los carbonos α influyen en los ángulos. (Alberts, y otros, 2015) y (Mckee & Mckee, 2014).
La hélice α es una estructura rígida en forma de varilla que se origina cuando una cadena polipeptídica se enrolla en una conformación
helicoidal dextrógira. Se forman enlaces por puente de hidrógeno en grupo N-H de cada aminoácido y grupo carbonilo del aminoácido que se encuentra cuatro residuos más adelante.
Las láminas plegadas β se forman cuando se alinean dos o más segmentos de la cadena polipeptidica, uno al lado de otro. Cada segmento individual se denomina cadena β. En lugar de estar enrollada, cada cadena β se extiende por completo y se estabiliza por medio de enlaces por puentes de hidrógeno que se forman entre los grupos N-H y carbonilo del esqueleto polipetídico de cadenas
adyacentes. Existen dos tipos de estructuras de las láminas β: las paralelas donde los puentes de hidrógeno están dispuestos en la misma dirección y las antiparalelas, los enlaces de puentes de
hidrógeno se encuentran en direcciones opuestas; se pueden llegar a observar cadenas paralelas opuestas. Algunas proteínas van a tener
combinación de hélices α y lámina plegada β. Estos patrones se denominan estructuras supersecundarias o motivos estructurales. (Mckee & Mckee, 2014).

lustración 4 Estructuras supersecundarias
seleccionadas. Unidad α-α; barril β; unidad meandro β; unidades
βαβ.
La AMPK (activated protein kinase) en español conocida como proteína cinasa activada. Es una enzima trimérica, constituida por una subunidad α y subunidades β y γ. Es una enzima crucial en la
regulación del metabolismo energético celular, cuya actividad se encuentra regulada por diversos mecanismos, entre los cuales el principal es el aumento en la concentración intracelular de AMP.
Esta enzima cataliza la fosforilación de múltiples proteínas reguladoras de las vías metabólicas de lípidos y de hidratos de carbono, con lo que favorecen su catabolismo (Hernández-Puga, y
otros, 2020). Esta enzima participa en procesos de
producción de energía como la glucólisis, la oxidación de lípidos y la gluconeogénesis (Mckee &Mckee, 2014).
Considerando la diversidad de cada proteína dependiendo la combinación de aminoácidos en cada secuencia polipeptídica y sus campos de fuerza que formen, tendrán una estructura única,
con un programa extensible para la visualización interactiva y el análisis de estructuras moleculares se van analizar la estructura de la enzima AMPK.
MATERIALES Y MÉTODO
- Se entró a la base de artículos RCSB y se eligió una proteína de interés, identificando los 4 caracteres propios de la proteína. Se
utilizó el programa ChimeraX y con el comando “Open_monbe” espacio de comando y el código previamente
identificado, se comenzó a trabajar con la proteína. - Con la opción de control, Action, Color. Se colorea la proteína seleccionada.
- En la opción de Tools, Structural Analysys, Distance se calculan dos distancias siempre y cuando se haya seleccionado los
átomos que se desea calcular las distancias. - Seleccionando la molécula en la opción de Tools, Structural Analysys, H-Bonds se marcan los puentes de hidrógeno.
- En la opción de Tools, Structural Analysys, angles/torsion se marcan los ángulos, siempre que se selecciones tres átomos.
- En la opción de Tools, Structural Analysys, match maker se pueden contraponer dos proteínas.
- La reconstrucción de proteínas en Swiss Model, es necesario que estén en formato fasta y de ahí convertirlos en código ordenado con números.
- Una vez que Swiss Model reconstruye proteínas, es importante que sí aparecen dos modelos, guiarse por la barrita azul de
coverage, por lo general el modelo 1 es el que baraca más y se parece más en identidad, la barra coverage indica en
cuanto porcentaje se reconstruyó. Así como el QMEANDisCo Global, indica la calidad de la proteína que va de 0.5 a 1.
Siempre muestra dos colores, naranja y morado; el morado representa mayor calidad de la proteína.
RESULTADOS
Práctica 1: Elegir una proteína e irse
adaptando al programa
Figura 1. Seleccionar proteína de interés. Con ctl se elegía un aminoácido.
Práctica 2: marcar segunda estructura B plegada y
colorear las cadenas de distintos colores.
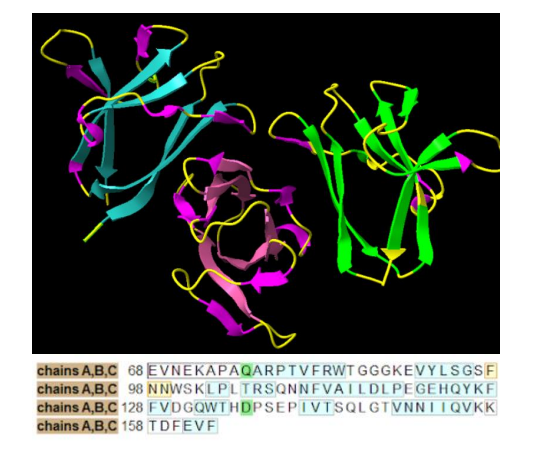
Figura 2. Seleccionar segunda estructura B e identificar las cadenas de la proteína
Práctica 3: identificación de aminoácidos.
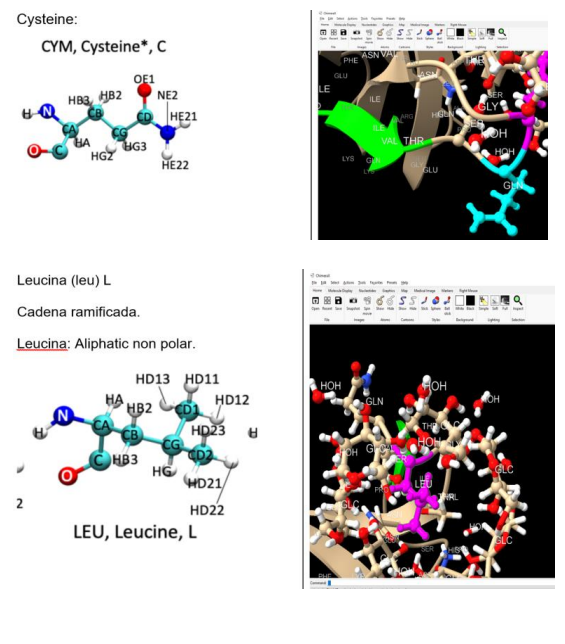
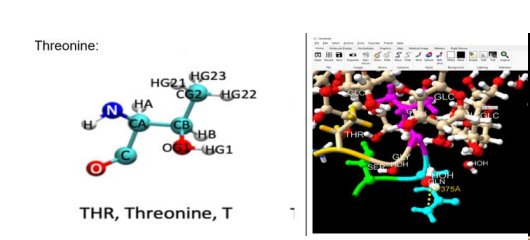
Figura 3. Identificación de aminoácidos en la proteína
Práctica 4: Calcular distancias
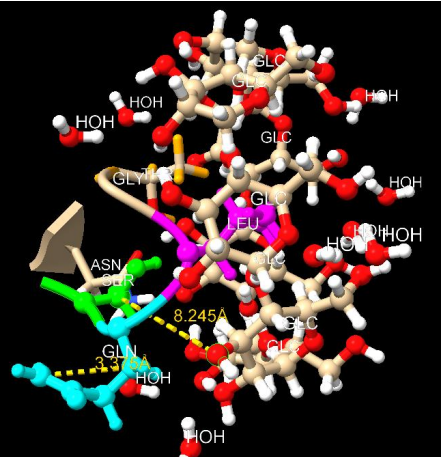
Figura 4: Calculo de distancias
Práctica 5: Calcular de fuerzas de Van
Der Waals y puentes de hidrógeno
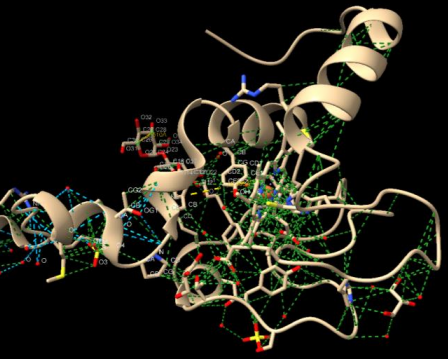
Figura 5. Fuerzas de Van Der Waals y puentes de
hidrógeno
Práctica 6: Calcular ángulos
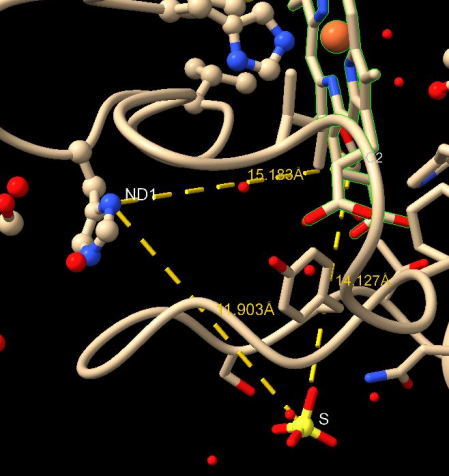
Figura 6. Cálculo de ángulos
Práctica 7: Contraponer dos modelos de la misma
proteína pero de diferente especie
CONCLUSIÓN
Se documentó la estructura de una proteína, aminoácido y enlaces, se ubicó la práctica teórica a partir de la visualización interactiva y se realizó el análisis de estructuras moleculares en programas
como ChimeraX y Swiss Model. A través de determinar distancias, campos de fuerza, aminoácidos se identifican las estructuras
supersecundarias Unidades beta-alfa-beta; meandro Beta como parte de las cadenas de la AMPK. Se identificaron ciertas variaciones en los loops de la AMPK de origen humano y mosca. Así como en la revisón mutaciones del AMPK en humanos. Lo visto en teoría coincide con el modelado de proteínas.
REFERENCIAS BIBLIOGRÁFICAS
- Mckee, T., & Mckee, J. R. (2014). Bioquímica Las bases moleculares de la vida. En T. Mckee, & J. R. Mckee, Bioquímica Las bases moleculares de la vida (págs. 110-141, 406- 408). México: McGRAW- HILL.
- Alberts, B., Johnson, A., Lewis, J., morgan, d., Raff, M., Roberts, K., & Walter, P. (2015).Biología Molecular de LA CÉLULA Sexta Edición. En B. Alberts, A. Johnson, J. Lewis, d. morgan, M. Raff, K. Roberts, & P. Walter,Biología Molecular de LA CÉLULA extaEdición (págs. 109-135). Estados Unidos de América: Garland Science.
- Lieberman, M., & Peet, A. (2018). Marks Bioquímica médica básica. En M. Lieberman, & A. Peet, Marks Bioquímica médica básica (págs. 80-86). Philadelphia: Wolters Kluwer.
- Hernández-Puga, G., Laguna-Maldonado, K. D., Gregg-García, R., Barrera-Zárate, G., LópezOrtiz, F. D., & Matuz-Mares, D. (2020). La AMPK como diana terapéutica del síndrome metabólico. Revista Médica del Instituto Mexicano del Seguro Social, 612- 621.
ANEXO:
Elegir una proteína e irse adaptando al
programa.
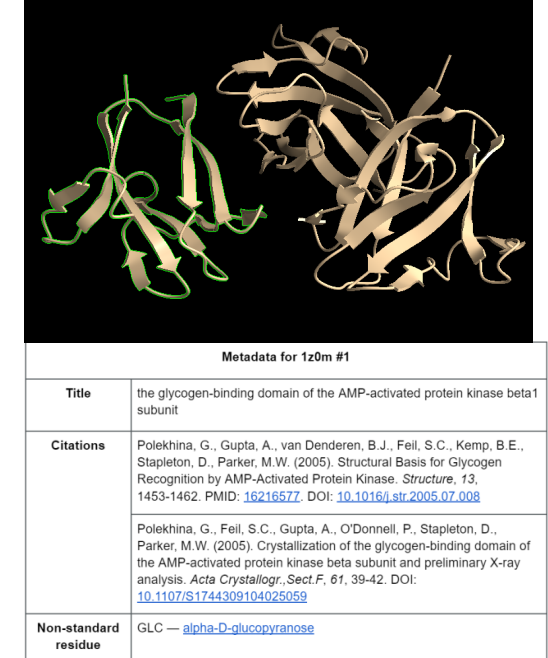
Figura Anexo 4.1 Seleccionar proteína de interés.
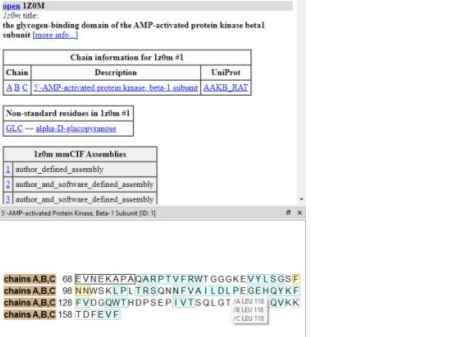
Figura Anexo 4.2 Ver secuencia de los aminoácidos
Práctica 2: marcar segunda estructura (beta
plegada)
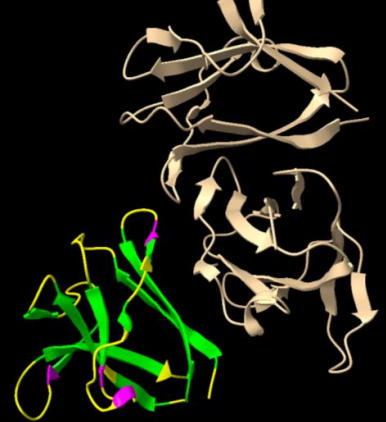
Figura Anexo 4.3 marcar segunda estructura (beta
plegada)
Identificar CADENA A
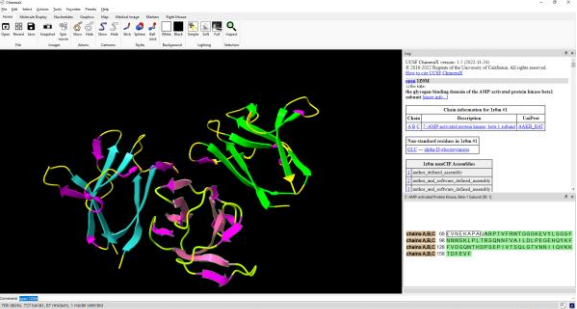
Figura Anexo 4.4 Identificar cadena A
Identificar CADENA b
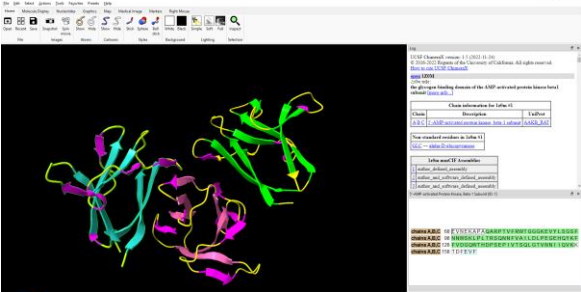
Figura Anexo 4.5 Identificar cadena B
Identificar CADENA c
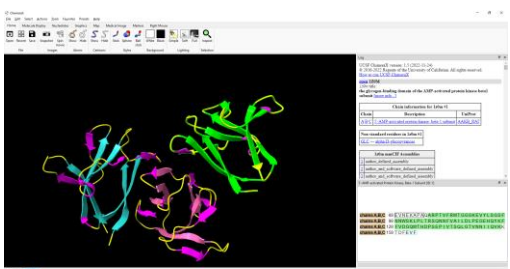
Figura Anexo 4.6 Identificar cadena C
Encontrar patrón de aminoácidos por cada
cadena
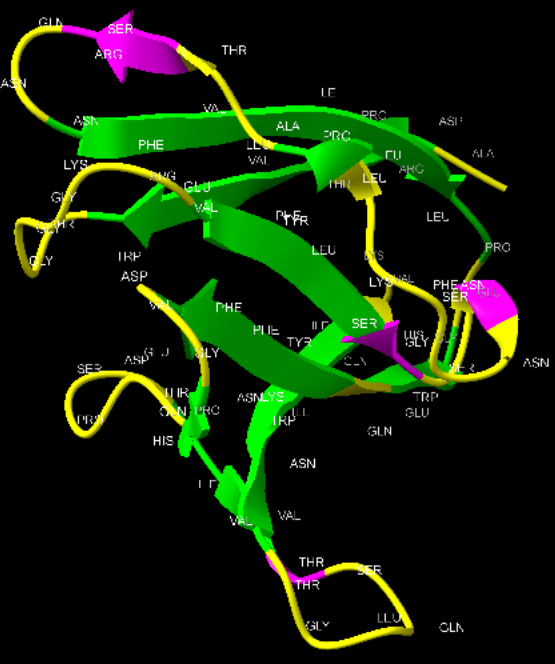
Figura Anexo 4.7 Encontrar patrón de aminoácidos por
cada cadena C
BUSCAR MUTACIONES
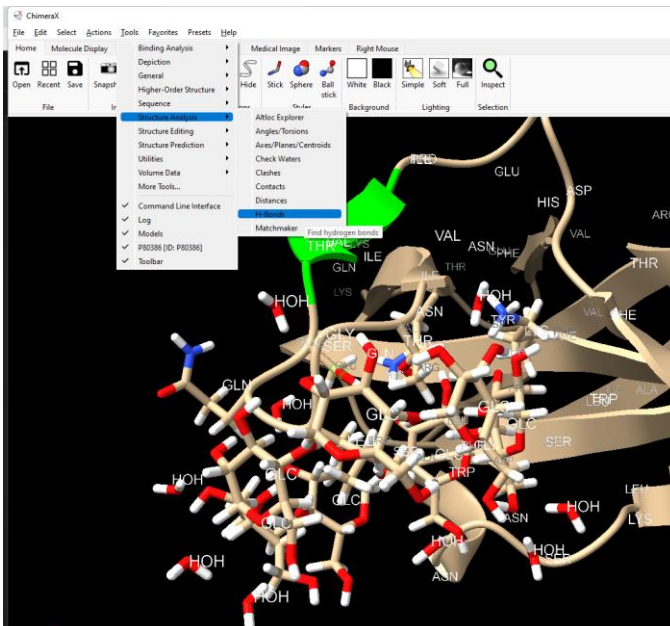
Figura Anexo 4.8 Buscar mutaciones Serina carbono
Beta
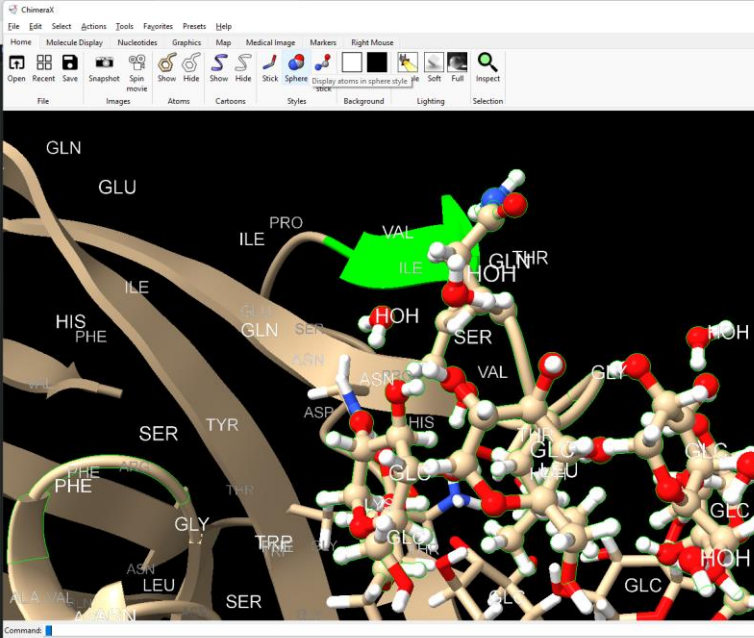
Figura Anexo 4.9 Buscar mutaciones Proline.
Práctica 3: identificación de aminoácidos
en la proteína seleccionada.

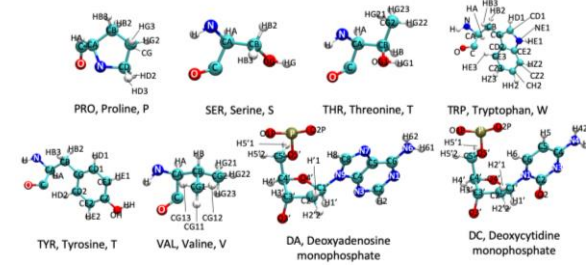
Figura Anexo 4.10 diagrama de estructuras usada para
la identificación de aminoácidos
4. Cálculo de distancias
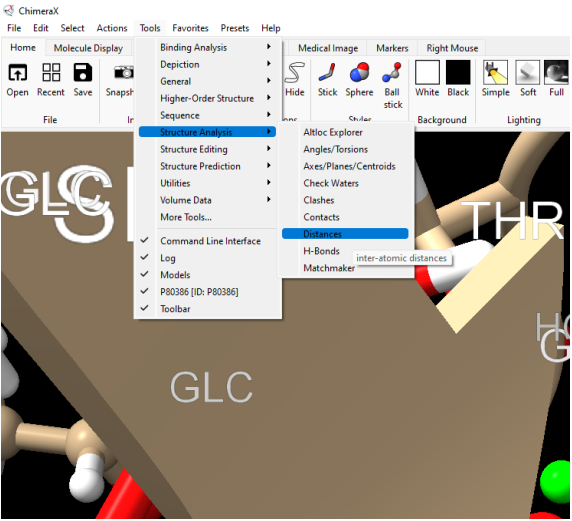
Figura Anexo 4.11 Tools / secuence analysis/ distance
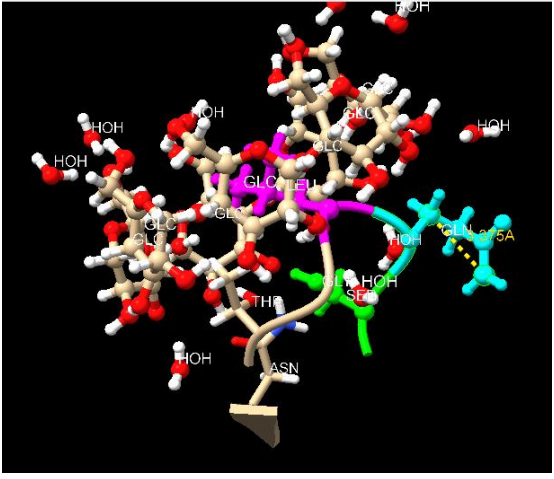
Figura Anexo 4.12 Calculo de distancias
5. Calcular de fuerzas de Van der Waals y
puentes de hidrógeno
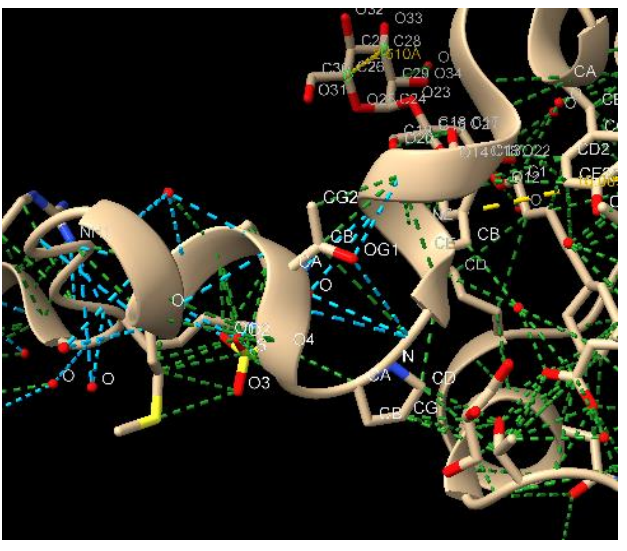
Figura Anexo 4.13 fuerzas de Van der Waals y puentes
de hidrógeno
6. Calcular ángulos

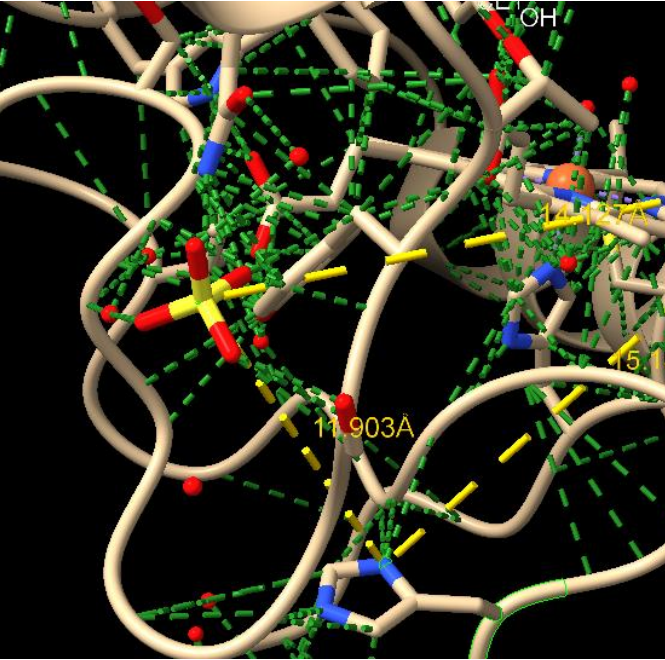
Figura Anexo 4.14 Calcular ángulos
Contraponer dos modelos de la misma
proteína pero de diferente especie.
Proteínas de interés: seleccionar la misma
proteína con la que se está trabajando en
diferentes especies
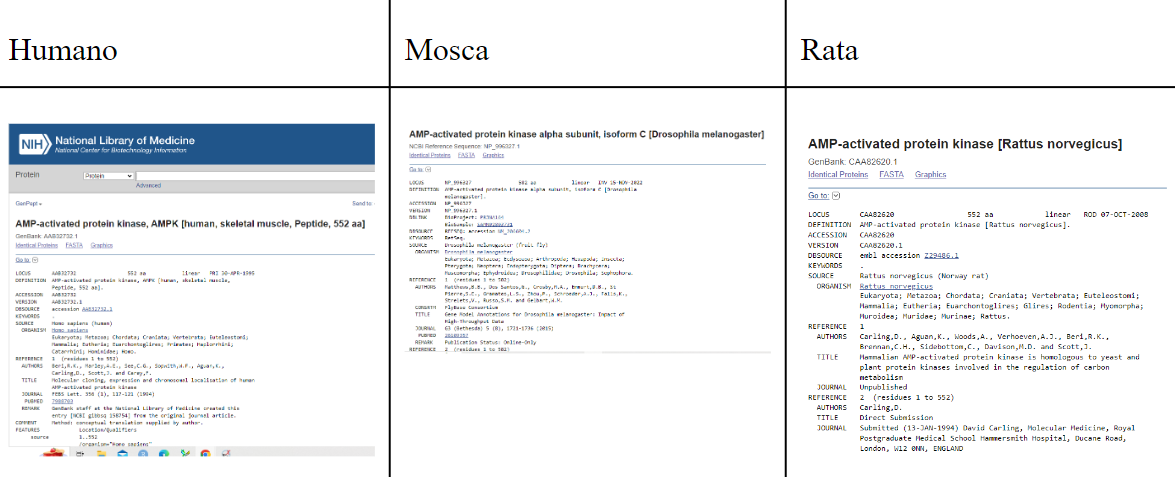
Figura Anexo 4.15 Buscar misma proteína diferentes
especies.